
Rede Corona-Ômica BR-MCTI identifica possível nova variante de SARS-CoV-2 emergente no Estado do Rio Grande do Sul
A Rede Vírus-MCTI comunica que a Rede Corona-Ômica BR-MCTI, através do Laboratório de Microbiologia Molecular (LMM), Universidade FEEVALE, Novo Hamburgo, Rio Grande do Sul (RS), sequenciou 26 genomas de SARS-CoV-2 de amostras de suabe nasofaríngeo, coletadas entre final de abril de metade de junho de 2021.
Do total de amostras, 20 são provenientes de viajantes que cruzavam a fronteira entre a Argentina e o RS, na cidade de Uruguaiana; quatro oriundas da Secretaria Municipal da Saúde de Porto Alegre (SMS) e duas que foram recebidas no próprio laboratório. As amostras foram selecionadas para o sequenciamento de alto desempenho através da plataforma Illumina MiSeq.
As sequências consenso dos 26 genomas completos, apresentando mais de 98% de cobertura, foram primeiramente submetidas a plataforma online Pangolin (https://github.com/hCoV-2019/pangolin), e caracterizadas como pertencentes a linhagem V.O.I B.1.1.28. No entanto, por tratar-se de uma linhagem atualmente pouco detectada, a análise de “variant calling” foi empregada nos arquivos FASTQ, com o objetivo de analisar possíveis novas mutações, em menor frequência. Como resultado, além das mutações características de B.1.1.28 (ORF1ab: L3930F e P4715L, proteína S: D614G e V1176F e proteína N: RG203KR), oito mutações não-sinônimas adicionais foram identificadas na ORF1ab (Q3777H, M3934I, Q3998R, L4182F, E4572D, RV4573QI, Q4576K, Y5601I), sete em S (G232A, I233L, N234P, N234K, T236S, V362E, E471Q), duas em N (P13L, D63E) e uma na ORF8 (A65S). Baseado nessas análises é possível que uma provável nova variante P.X tenha sido detectada, e que sua emergência esteja sendo observada em tempo real, em ração da metodologia aplicada.
Dentre as mutações adicionais observadas, seis chamaram atenção devido a frequência total em que apareceram nas amostras e que parecem ser assinaturas da provável nova variante P.X emergente, especialmente N234P e E471Q, na proteína S, e, M3934I e L4182F, na ORF1ab. É importante salientar que as mutações características de P.1 e P.2 não foram encontradas em nenhum dos genomas gerados neste estudo. Os resultados completos podem ser observados na Tabela 1.
Foi realizado, também, o alinhamento e a árvore filogenética dos 26 genomas completos, com sequências representativas de outras linhagens, assim como a sequência referência de Wuhan (hCoV-19/Wuhan/WIV04/2019). A filogenia (Maximum Likelihood, GTR+I, 1000 bootstraps) foi realizada através da plataforma online IQtree v2.1.2, e mostrou que as sequências claramente agrupam-se em um clado separado das demais, corroborando os resultados anteriores de uma provável nova variante em emergência, mais relacionada à B.1.1.28 (Figura 1).
A constante descoberta de novos genomas e possíveis novas linhagens, assim como o monitoramento de novas mutações são de especial relevância, considerando o impacto causado por outras linhagens como B.1.1.7 (Alpha), B.1.351 (Beta), P.1, e B.1.617.2 (Delta), no Reino Unido, África do Sul, Brasil e Índia, respectivamente. Todos os dados estão sendo disponibilizados em bases de dados públicos nacionais (Corona-Ômica.BR – MCTI) e internacionais (GISAID) com a posterior submissão do trabalho ao periódico científico.
Tabela 1. Análise de “variant calling” realizada nos arquivos de reads (FASTQ). As frequências máximas e mínimas representam o maior e menor percentual em que uma mutação foi detectada em uma dada amostra. A frequência total representa o total de amostras em que uma dada mutação foi identificada.
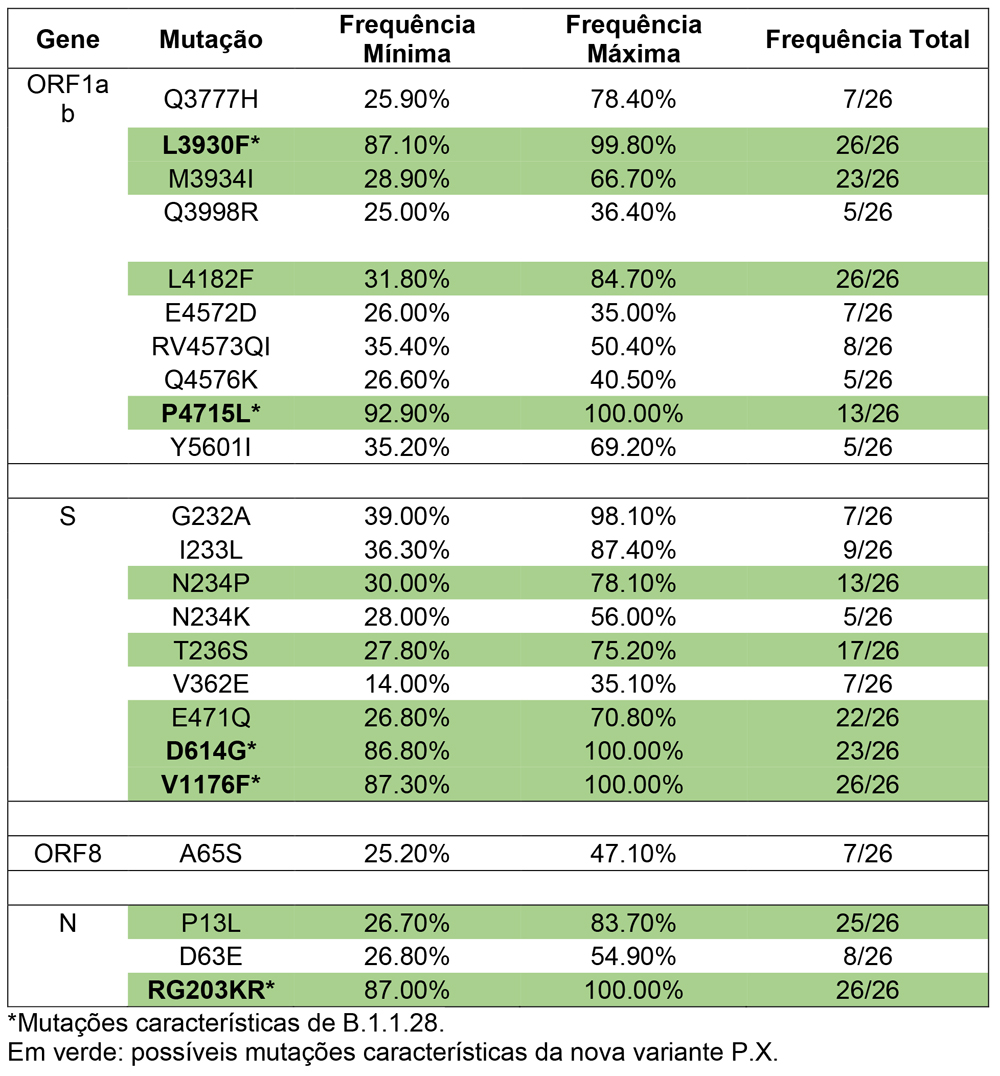

Figura 1. Análise filogenética do genoma completo de SARS-CoV-2. Na esquerda, uma filogenia global gerada pela plataforma online Nextclade. Na direita, análise filogenética Maximum Likelihood, GTR+I, 1000 bootstraps, gerada no servidor online IQ-TREE v.2.1.2. Marcadas na caixa verde, as sequências geradas neste estudo.
Fonte: https://www.gov.br/mcti

